
Mapping Protein–Protein Interactions by Mass Spectrometry
Mapping Protein–Protein Interactions by Mass Spectrometry: A Comprehensive Guide to Modern Interactomics
Introduction
Protein–protein interactions (PPIs) form the molecular backbone of all cellular processes. From signal transduction and chromatin remodeling to immune responses and metabolic regulation, proteins rarely act alone. Instead, they assemble into dynamic, context-dependent complexes that define cellular function.
Deciphering these interaction networks collectively known as the interactome is essential for understanding both normal physiology and disease mechanisms. Over the past two decades, mass spectrometry (MS)-based proteomics has emerged as the most powerful and versatile platform for large-scale PPI mapping.
Unlike traditional binary interaction assays, MS-based approaches enable system-wide, unbiased, and quantitative characterization of protein complexes in their native cellular environments.
The Biological Importance of Protein–Protein Interactions
Protein interactions can be classified into several categories:
Stable Interactions
Found in multi-subunit complexes (e.g., ribosomes, proteasomes)
Long-lived and structurally defined
Transient Interactions
Occur during signaling cascades
Weak and short-lived
Conditional Interactions
Depend on cellular states (e.g., phosphorylation, stress, ligand binding)
Disruption of PPIs is a hallmark of many diseases:
Cancer: aberrant signaling complexes
Neurodegeneration: protein aggregation and misfolding
Infectious diseases: host–pathogen interaction hijacking
Thus, mapping PPIs is not only descriptive but also mechanistically and therapeutically critical.
Why Mass Spectrometry Dominates PPI Analysis
Mass spectrometry offers a unique combination of features:
✔ Sensitivity & Dynamic Range
Modern MS instruments detect proteins across several orders of magnitude in abundance
✔ Specificity
Peptide-level identification ensures precise protein assignment
✔ Throughput
Thousands of proteins can be analyzed in a single experiment
✔ Quantitative Capability
Label-free or isotope labeling approaches allow interaction strength measurement
✔ Structural Insight
When combined with cross-linking, MS provides spatial constraints
These advantages position MS as the central technology in interactomics.
Core Mass Spectrometry-Based PPI Mapping Strategies
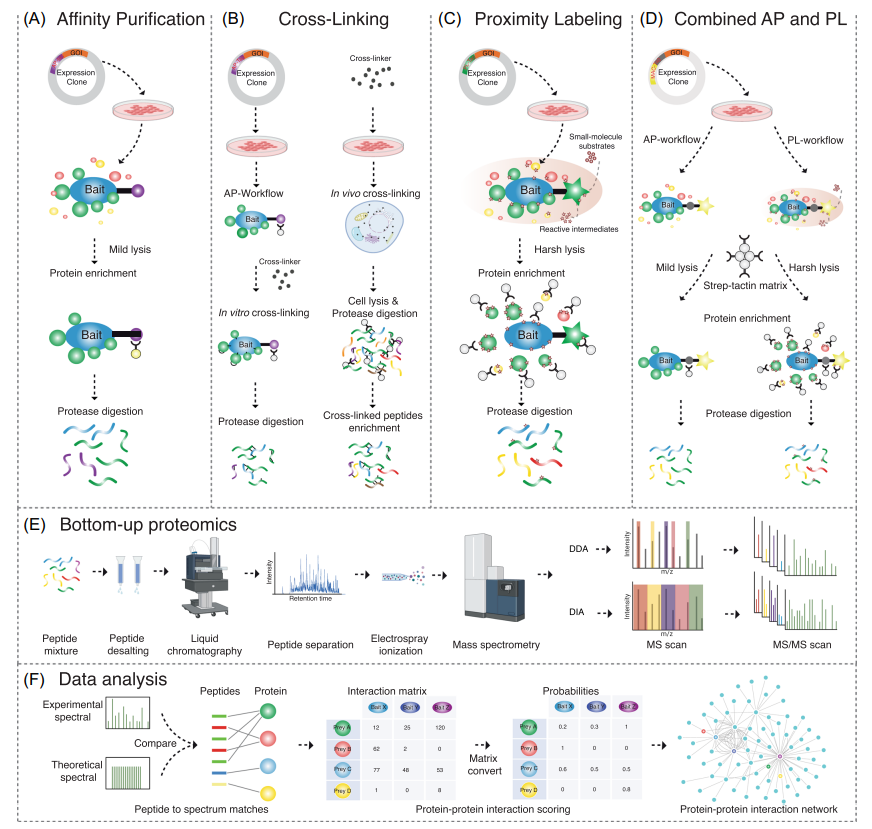
FIGURE 1 MS‐based approaches to studying interactomes. (A) Affinity tags are used in affinity purification (AP). Protein of interest (POI) that
has been affinity tagged can be produced transiently or stably in selected cell lines. Subsequently, matrix conjugated to antiaffinity tag antibodies are
added so that the tag fused POI and its interactors can be selectively enriched. (B) Cross‐linking can be done in vitro after purifying protein complexes
or in vivo with intact cells. The cross‐linked proteins are digested to produce cross‐linked peptides, which are then enriched before mass spectrometry
(MS) analysis. (C) The proximity labeling (PL) procedure. An enzyme is genetically fused to the POI and expressed in the cell line of choice. In vivo
labeling is accomplished by introducing substrate into the cells, which converts these molecules into reactive intermediates for PL. Labelled proteins
can be enriched. (D) Combination of AP and PL. Depending on the culture state and lysis buffer combination, MAC‐tagged POI can be utilized for
both AP and PL procedures. The same matrix is used to enrich protein complexes. (E) In bottom‐up proteomics, peptides derived from proteolytic
digestion are first desalted to remove salts that can interfere with subsequent analyses. The cleaned peptide mixture is then loaded onto an liquid
chromatography column for liquid chromatography, followed by electrospray ionization. Ionized peptides are analyzed using two primary MS
strategies: data‐dependent acquisition (DDA), where ions are scanned and the most abundant peptides are chosen for MS/MS scans, and data‐
independent acquisition (DIA), where all peptides within a set mass range are systematically fragmented. These approaches yield tandem mass
spectra for peptide identification and inference of associated proteins. (F) Data analysis begins with the comparison of experimental spectra against a
theoretical database to establish peptide–protein matches, which then form an interaction matrix. Interaction probabilities are statistically scored to
recognize high‐confidence interactions (HCIs). These HCIs, extracted from filtering processes, are used to construct a protein–protein interaction
network, elucidating the intricate web of protein interactions within the cellular environment.
1. Affinity Purification–Mass Spectrometry (AP-MS)
Principle
A “bait” protein is isolated along with its interacting partners (“prey”) using affinity-based methods. The purified complex is then analyzed via LC-MS/MS.
Experimental Workflow
-
Genetic tagging (e.g., FLAG, HA, GFP) or antibody-based capture
-
Cell lysis under native conditions
-
Affinity purification
-
Stringent washing to remove non-specific binders
-
Enzymatic digestion (trypsin)
-
LC-MS/MS identification
Strengths
High specificity
Suitable for stable complexes
Scalable for proteome-wide studies
Limitations
Loss of transient/weak interactions
Potential artifacts from overexpression
Advanced Variants
Tandem affinity purification (TAP)
Quantitative AP-MS (using SILAC or TMT)
2. Proximity Labeling Approaches
These methods overcome AP-MS limitations by capturing proteins in close spatial proximity rather than requiring stable binding.
Key Technologies
BioID (biotin ligase-based labeling)
APEX (engineered peroxidase labeling)
Mechanism
-
Bait protein fused to an enzyme
-
Enzyme generates reactive labeling species
-
Nearby proteins are covalently tagged (e.g., biotinylated)
-
Labeled proteins are purified and identified by MS
Advantages
Captures transient and weak interactions
Works in live cells
Spatially resolved interactomes
Limitations
Labeling radius may include false positives
Requires careful controls
3. Cross-Linking Mass Spectrometry (XL-MS)
Concept
Chemical cross-linkers covalently connect interacting amino acid residues, preserving protein interactions during analysis.
Workflow
-
Apply cross-linking reagent (e.g., DSS, BS3)
-
Digest proteins into peptides
-
Enrich cross-linked peptides
-
Analyze via high-resolution MS
Unique Advantages
Provides distance constraints (structural insight)
Maps interaction interfaces
Useful for modeling protein complexes
Challenges
Complex data analysis
Low abundance of cross-linked peptides
4. Co-Fractionation Mass Spectrometry (CoFrac-MS)
Principle
Protein complexes are separated by biochemical fractionation:
Size exclusion chromatography
Ion exchange
Density gradients
Proteins that co-elute across fractions are inferred to interact.
Strengths
No genetic manipulation required
Suitable for endogenous complexes
Scalable to proteome-wide mapping
Weaknesses
Indirect inference of interactions
Requires computational modeling
Quantitative Proteomics in PPI Analysis
Quantification is essential for distinguishing true interactors from background.
Common Approaches
Label-Free Quantification (LFQ)
Based on peptide intensity
Simple but sensitive to variability
SILAC (Stable Isotope Labeling)
Incorporates heavy amino acids in vivo
High accuracy for comparative studies
Tandem Mass Tags (TMT)
Multiplexing capability
High throughput
Quantitative data enables:
Interaction strength measurement
Dynamic interaction studies
Condition-specific interactome analysis
Computational Analysis of PPI Data
MS-based PPI datasets are large and complex, requiring advanced bioinformatics:
Key Steps
-
Peptide identification (search engines like Mascot, Sequest)
-
Protein inference
-
Contaminant filtering
-
Statistical scoring (e.g., SAINT, MiST)
-
Network reconstruction
Challenges
False positives from non-specific binding
Batch effects
Missing data
Visualization Tools
Cytoscape
STRING database integration
Experimental Design Considerations
To ensure high-quality PPI data:
Controls
Negative controls (empty tag)
Mock purifications
Replicates
Biological and technical replicates improve confidence
Stringency
Optimize washing conditions to reduce background
Tag Selection
Avoid interfering with protein function
Applications of MS-Based Interactomics
Systems Biology
Reconstruction of protein interaction networks
Identification of functional modules
Drug Discovery
Targeting protein interaction interfaces
Identification of druggable complexes
Disease Mechanisms
Mapping altered interactomes in cancer
Studying neurodegenerative protein aggregation
Host–Pathogen Interactions
Understanding viral hijacking of host machinery
Identifying therapeutic targets
Emerging Trends in Interactomics
Single-Cell Proteomics
Enables PPI analysis at cellular resolution
Critical for heterogeneous tissues
Multi-Omics Integration
Combining:
Proteomics
Transcriptomics
Genomics
→ Provides a holistic view of cellular systems
Improved Instrumentation
Higher resolution MS
Faster acquisition speeds
Enhanced sensitivity
Conclusion
Mass spectrometry has fundamentally transformed the study of protein–protein interactions, enabling researchers to move from single interactions to complex, dynamic interaction networks.
Each approach AP-MS, proximity labeling, XL-MS, and co-fractionation offers complementary insights. When combined with quantitative proteomics and computational analysis, these methods provide a powerful framework for understanding cellular biology and disease mechanisms.
As technologies continue to evolve, MS-based interactomics will play a central role in precision medicine, drug discovery, and systems biology.
Recent Posts
-
Types of Plasmid
Types of Plasmid: A Complete Scientific Guide Introduction to Types of Plasmid Understanding the typ …8th Apr 2026 -
Spectrophotometer Principle
Spectrophotometer Principle: A Complete Scientific Guide Introduction to Spectrophotometer Principle …8th Apr 2026 -
Types of Media in Microbiology
Types of Media in Microbiology: Complete Guide for Laboratory Use Introduction to Types of Media in …6th Apr 2026